

同義詞
遺傳性感覺自律神經病變1A型
(Hereditary Sensory and Autonomic Neuropathy type 1A; HSAN1A)
遺傳性感覺自律神經病變1型
(Hereditary Sensory and Autonomic Neuropathy type 1; HSN1)
遺傳性感覺神經病變1型
(Hereditary Sensory Neuropathy 1; HSN1)
病因
HSAN1A 是遺傳性(基因性) 周圍神經疾病。病因爲顯性體染色體SPTLC1基因突變。這個基因提供製造SPT轉移酶長鏈第一亞單元(Serine Palmitoyltransferase Long Chain Subunit 1)的訊息。SPT轉移酶共有3個亞單元, 分別由SPTLC1,SPTLC2, SPTLC3三個基因提供訊息而構成。SPTLC1 位於第九號染色體內,SPTLC2位於第14號染色體,SPTLC3位於第20號染色體內1-5。
其中SPTLC1 和 SPTLC2基因已被科學家發現若具突變,則可能造成遺傳性感覺自律神經病變(Hereditary Sensory Autonomic Neropathy, 英文簡稱HSAN)。SPTLC2基因的突變造成HSAN, 1C型4。 SPT轉移酶的正常功能為催化L型絲胺酸(L-Serine) 和軟脂醯輔酶A (Palmitoyl-CoA)的凝結, 這也是首先合成神經鞘脂(Sphingolipids)的第一步,同時也是速度控制的步驟6。神經鞘脂為人體細胞膜組成的重要元素。
SPT轉移酶的正常功能是協助絲胺酸(人體內胺基酸的一種)和軟脂醯輔酶A(Palmitoyl-CoA)的結合,此生化反應的產物為鞘氨醇(Sphinganine, 簡稱SA)。這是一連串產生鞘脂的第一個步驟。然而突變後的SPT轉移酶改變了其特性,它除了和原本的L型絲胺酸反應,也和L型丙氨酸(L-alanine)反應,形成其產物 1-脱氧鞘氨醇(1-deoxy-sphinganine, 簡稱1-deoxy-SA) 。某些SPT的突變體甚至與L型甘胺酸(L-Glycine)反應,形成1-脫氧甲基鞘氨醇(1-deoxymethyl-sphinganine, 簡稱 1-deoxymethyl-SA)。1-脱氧鞘氨醇和1-脫氧甲基鞘氨醇皆為脫氧鞘脂。研究人員發現脫氧鞘脂和正常鞘脂的生化結構有些許不同(它們皆缺乏了鞘氨醇中C 1的氫氧群)因此它們無法參與下一步驟的正規生化反應形成鞘脂,也無法在人體內被降解,因此囤積於細胞中,也存在於血漿內6。研究人員於觀察中發現脫氧鞘脂對神經具有毒性,並且大量堆積於坐骨神經,但幾乎不囤積於腦和脊髓,因此它損害周圍神經系統而不損害中樞神經系統。周圍神經系統中的感覺神經,運動神經,及自律神經皆受到影響。實驗發現,脫氧鞘脂對感覺神經的損害最為嚴重,其次為運動神經,對自律神經的影響似乎較不顯著6,13,16。
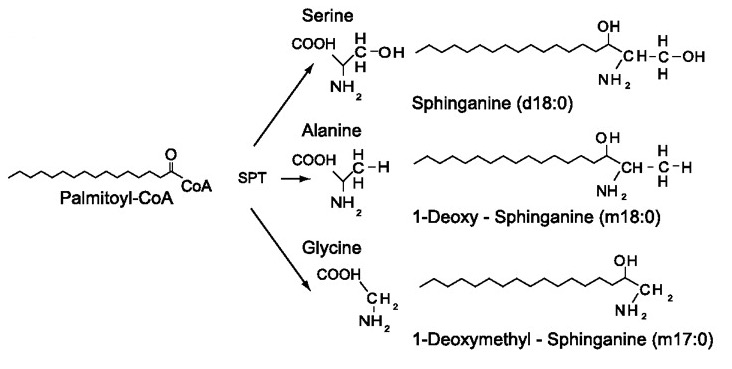
突變後的SPT轉移酶與丙氨酸(Alanine)和甘胺酸(Glycine)結合,其產物 1-脱氧鞘氨醇(1-deoxy-sphinganine) 和1-脫氧甲基鞘氨醇(1-deoxymethyl-sphinganine)在化學結構上並不具鞘氨醇在C1的氫氧群(OH),因此無法參與下一階段的生化反應成為鞘脂。(圖取自 Penno et al. 2010)
進一步的研究發現脫氧鞘脂損害神經細胞是透過損害細胞內俗稱能量工廠的粒線體。低濃度的脫氧鞘脂對粒線體並不造成傷害,然而高濃度的脫氧鞘脂使其形狀和結構改變,甚至使它成為碎片。當濃度高達1 μM 時,細胞死亡 ⁹ 。
(圖/ 美國人類基因體國家研究中心提供;National Human Genome Research Institute.
https://www.genome.gov/genetics-glossary/Mitochondria)
正常健康的人體內也會形成脫氧鞘脂,但是含量相當低,大約 0.1-0.3 μM,這並不會影響細胞。然而 HSAN1A的患者體內的脫氧鞘脂含量可高達1.2 μM。高量的脫氧鞘脂也存在於第二型糖尿病患者和代謝症候群患者中 ⁹。
症狀
HSAN1A 影響周圍神經系統。影響部位主要在腳和手,較嚴重病例可能延伸至大腿和上臂。感覺神經,運動神經,和自律神經皆可能受到損害,以感覺神經影響最為嚴重。患者在發病初期在下肢末端會有一段過渡期的痛覺過度(Hyperpathia, 又稱感覺過度,感覺異常)之後皮膚就會失去對溫度和疼痛的感覺。一段時間之後,感覺失去的症狀也會發生在手的部位。因感覺不到痛,患者經常會受傷或燙傷而不自知 10,11,13。如果傷口不適當處理,容易引起重複感染和長期潰瘍而導致骨髓炎的發生,並且面臨截肢的風險。隨著病情進展,患者在手和腳會有麻刺感,灼熱感,和類似電擊性的疼痛。之後這些症狀可能擴散到大腿和上臂。感覺失去也可能發生在腹部,從中間部位開始,逐漸擴散至兩側和上胸。關節的位置感和震動感也受影響但較不嚴重,並且多半發生於皮膚失去痛覺和分辨溫度的能力之後10。

大部分患者的運動神經功能也受到不同程度的損害。若是發生,患者的手腳部位會感到肌肉無力和萎縮。腳踝無力,無法踮腳尖,無法用腳跟走路也是常見的情況。較嚴重的病例肌肉萎縮也發生於小腿,甚至擴散到大腿10,11 。相同症狀發生於上肢時,患者會感到手指的靈活度下降,做精細的事情會有困難,比如扣扣子,使用針線,綁鞋帶,等等。上肢的症狀多半在下肢的初期症狀開始的數年後發生。
HSAN1A 也可能造成足部畸型,比如夏兒哥足,高弓足,扁平足,錘狀趾10,11。到了疾病的中後期,患者可能在行動上需要使用輔助器具,比如腳架,步行器,輪椅,或電動代步車,等等。
較少患者有自律神經的損害。目前為止,腸胃道症狀(腹部疼痛,便秘,拉肚子,脹氣,體重減輕) 心跳問題,低血壓,流汗異常(流汗過多,過少,或是無汗)血管舒縮功能異常,等病例報告 10,¹¹,¹²。皮膚方面,HSAN1患者在患部的皮膚較細緻13。眼睛方面,可能引起瞳孔收縮異常(強直瞳孔, tonic pupil)12。有些變異位點有青少年白內障的記載 ⁸,15。最新的研究顯示高量的脫氧鞘脂可能造成黃斑部病變第二型(Mac Tel type 2)¹⁴ 。
HSAN1A 也可能造成聽力受損,如果發生,則多半發生於中年之後。智力基本上不受影響。關於壽命,曾有一項對於6個患有HSAN1A的英國家庭的研究顯示,患有HSAN1A的家庭成員的平均壽命為67歲,和英國一般大眾的平均歲數相比提早了些,推測其原因是來自此病的併發症 10。除此之外,有一突變位置S331,全球目前共發現極少數患者有其特別嚴重的症狀,其中也有於20歲左右病故的個案 15。
發病時間
大部分患者的發病時間介於10-60歲之間¹² 。較少患者開始於兒童期。兒童期發病的患者通常症狀較嚴重8,10,15。
此病的發病年齡、症狀、和嚴重程度因人而異,在不同的家族之間或甚至相同家族不同成員中的表型不盡相同。一般來說,患者的表型與突變的類型和患者血漿內的脫氧鞘脂含量有關 ⁹。較高量的脫氧鞘脂傾向於對神經造成較多損害並且造成較嚴重的症狀。過度飲酒也可能使症狀加劇⁶。此外,研究人員推測應有其他調控基因或後天因素對患者的表型產生影響10。
恰克. 瑪俐. 杜斯氏症(C.M.T.) 與 HSAN1A 的關係
恰克. 瑪俐. 杜斯氏症 (英文名 Charcot.Marie.Tooth neuropathy, 簡稱 CMT) 又稱腓骨肌萎縮症,為一群的遺傳性(基因性) 神經性疾病。病理特徵是緩慢並且進行性的損害周圍神經中的運動和感覺神經功能,主要影響部位為四肢。CMT患者的腳和手會出現肌肉無力/萎縮,對溫度和疼痛的感覺減退。目前為止已發現至少超過120個基因與CMT有關,意即這120個基因當中的任何一個基因若發生致病突變,則造成CMT的相關症狀發生。CMT損害運動和感覺神經,因此又稱為遺傳性運動和感覺神經病變 (Hereditary Motor Sensory Neuropathy, HMSN)。遺傳性感覺和自律神經病變1A型,此病從名稱上看起來只損害感覺和自律神經,然而,如同上述陳列的症狀,運動神經也受影響。HSAN1A患者若呈現感覺和運動神經症狀並且無自律神經功能損害或較不明顯,臨床上會得到遺傳性運動感覺神經病變, 即CMT的診斷 10,17。近幾年,因SPTLC1 和 SPTLC2 基因突變所導致的神經肌肉疾患不只被歸類於HSAN1, 也被歸類於CMT第二型(軸索型), 又稱 HMSN 第二型 12,18。

診斷
診斷可包括下列檢查: 基因檢查、詳細的臨床評估和家族史詢問、神經傳導速度檢查、肌電圖檢查、自律神經功能測試¹2 。抽血檢測HSAN1A的生物標記-脫氧鞘脂,此血液分析可在瑞士蘇黎世大學實驗室協助下完成6,16。
疑似HSAN1A的患者在診斷上常面臨挑戰,因為HSAN1A的症狀像極了CMT的某些亞型,比如CMT2B (rab7基因突變造成)因此診斷需要依靠基因篩檢。然而基因檢查並不是在每個國家都普及,也不是人人在經濟上都負擔得起。即使患者有機會做基因檢測, 如果篩檢出的突變結果顯示突變意義不明確 (Variant of Unknown Significance)(意指不確定屬於良性突變或是致病突變)就會需要分析血漿中的脫氧鞘脂含量來判定,然而這個檢查並不是隨處可以做,特別是生物樣本的國際運輸可能遇到困難,因此篩檢出來的突變到最後仍然無法確定它是否致病。
盛行率
HSAN1A的發生率大約從10萬人當中1人至100萬人中2人11,12。
發生率的計算相當困難,因為不少患者的症狀只有感覺功能的損害和肌肉無力/萎縮,因此診斷為CMT。SPTLC1之前並未列於CMT篩檢的基因當中,因此患者即使做了基因檢查,也可能錯過此基因而檢測不出來。
治療
目前並沒有痊癒的方法。然而,值得一提的是口服L型絲胺酸療法。由於突變後的SPT轉移酶不只和絲胺酸結合,也和丙氨酸結合,研究員就思考如果可以增加絲胺酸/丙氨酸在人體內的比例,讓SPT突變體有較多的機會和絲胺酸結合並且較少的機會和丙氨酸結合,是否就可以降低脫氧鞘脂的生成,進而減緩疾病的進展?
研究人員在實驗中發現,在攜有SPTLC1 C133W 基因突變的基因鼠膳食中增加10%的L形絲胺酸,僅於2-4天以內,1-deoxySA的含量大幅降低; 而相較於沒有服用絲胺酸的基因鼠,持續10個月服用絲胺酸(膳食中增加10%的絲胺酸)的基因鼠在機械性敏感度和運動的表現上都有改善13。
基於這樣成功的結果,研究人員在2010年做了首次的人體試驗。這是一個短期10週的試驗,共有14位具有SPTLC1 C133Y 突變的HSAN1A患者參與。患者分為兩組,分別給與低劑量200mg/kg/day (根據受試患者體重每公斤給予200毫克的絲胺酸/每日)和高劑量400mg/kg/day的絲胺酸。結果在一個月之內,兩組受試者血漿中的脫氧鞘脂 (1-deoxySL)含量均下降,高劑量的降低幅度較大。在此次的研究中,並沒有測量運動和感覺的表現,因為此次的研究焦點主要爲測量患者體內的生化反應。臨床表現並不被認為會獲得改善,因為患者多年的神經受損不太可能在短期的治療時間內得到緩解。儘管如此,部份受試病患表示手部的麻刺感,生理期的疼痛感增加,並且皮膚的堅實度,頭髮和指甲的生長速度也快速些13。
研究團隊於2017 年完成了為期兩年(96週)對於HSAN1A患者進行口服絲胺酸臨床試驗(ClinicalTrial.gov identifier NCT 01733407) 共18名HSAN1A患者參與。患者隨機被分為同等人數的兩組(9人與9人)其中一組被給予絲胺酸,另一組給予安慰劑。發配的絲胺酸和安慰劑皆為粉末狀,外觀和味道完全相同(安慰劑對照)試驗人員與受試患者兩方皆不知發配的粉末為絲胺酸或安慰劑(雙盲)第一年絲胺酸組的受試者每日根據體重每公斤給予400毫克的絲胺酸,另一組給予相同劑量的安慰劑。第二年,兩組都服用絲胺酸,劑量與第一年相同。共16位受試者完成試驗。 試驗結果顯示絲胺酸組連續兩年( 於第48週和96週複診測量時)在感覺和四肢的力氣方面得到改善(使用Charcot-Marie-Tooth Neuropathy Score version 2, CMT神經病變評分第二版來測量)而安慰劑組在第一年(48週測量時)症狀退步,在第二年(96週測量時)症狀上獲得改善。受試者血漿中的脫氧鞘脂的變化方面,絲胺酸組在第一年(第48週)結束時1-deoxySA 降低了60%,而安慰劑組則升高了9%。安慰劑組於第二年開始服用絲胺酸,在第二年結束時(第96週)安慰劑組的1-deoxySA 降低了66%。在此次的臨床試驗中沒有嚴重的副作用發生(輕微至中度的不良反應請參考文獻第e366頁*)此臨床試驗做出以下結論:高劑量的絲胺酸治療似乎是安全的,並且對於HSAN1A病患的疾病進展具潛在的減緩效果16。
(註:上述臨床實驗的受試者人數少,任何想服用絲胺酸的HSAN1A患者請與醫師充分溝通,於醫師指示下服用)
致病突變位點
目前為止,已證實並且發表於文獻的SPTLC1致病變異位點有: SPTLC1 p.133W, C133Y, C133R, C133F, V144D, S331F, S331Y, A352V¹,2,7,8,19。
遺傳模式
HSAN1A遺傳模式為體染色體顯性遺傳
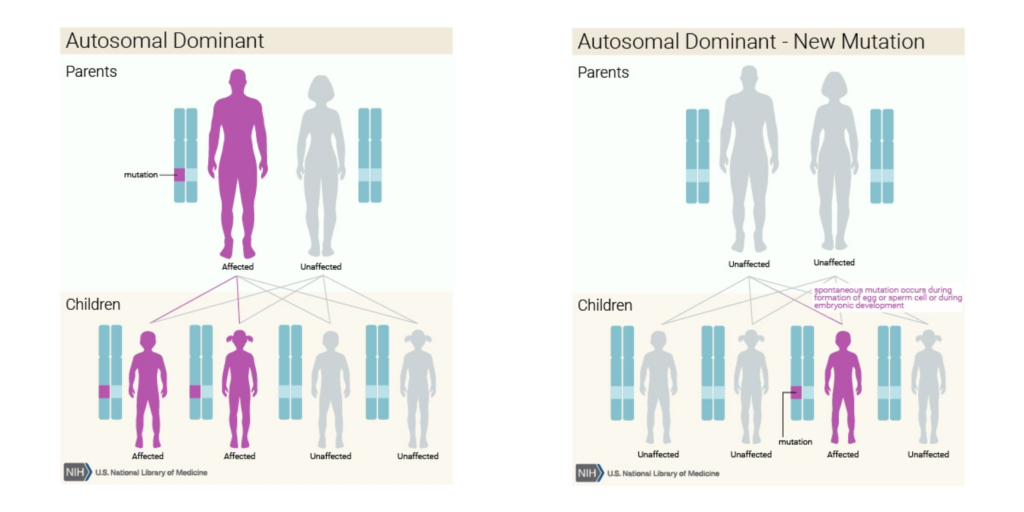
或親代皆為健康,子代的個體突變(de novo)。(右圖)
圖片來源:美國國家醫學圖書館
最後小叮嚀: 登載在美國衛生局(National Institutes of Health)網站的文獻表示若有以下症狀符合,即要懷疑自己的疾病是否由SPTLC1基因突變造成。1.病情開始於感覺神經病變後來演變為運動和感覺神經軸索病變。2.手和腳的末端即使受傷也不感覺疼痛、神經性關節病變(Charcot joints), 有時甚至需要截肢。3.遠端肌肉無力漸至擴散到四肢近端肌肉。4.病情到了某一階段,有電擊似的疼痛發生。5.有些病人有感覺神經聽力損失。6.有體染色體顯性遺傳的家族史(有極少數已證實的HSAN1病患是個體新的突變(de novo mutation),無家族史的12。
資料來源
1 Bejaoui K, Wu C, Scheffler MD, Haan G, Ashby P, Wu L, de Jong P, Brown RH Jr. SPTLC1 is mutated in hereditary sensory neuropathy, type 1. Nat Genet. 2001 Mar;27(3):261-2. doi: 10.1038/85817. PMID: 11242106.
2 Dawkins JL, Hulme DJ, Brahmbhatt SB, Auer-Grumbach M, Nicholson GA. Mutations in SPTLC1, encoding serine palmitoyltransferase, long chain base subunit-1, cause hereditary sensory neuropathy type I. Nat Genet. 2001 Mar;27(3):309-12. doi: 10.1038/85879. PMID: 11242114.
3 Hornemann T, Penno A, Rütti MF, Ernst D, Kivrak-Pfiffner F, Rohrer L, von Eckardstein A. The SPTLC3 subunit of serine palmitoyltransferase generates short chain sphingoid bases. J Biol Chem. 2009 Sep 25;284(39):26322-30. doi: 10.1074/jbc.M109.023192. Epub 2009 Aug 1. PMID: 19648650; PMCID: PMC2785320.
4 Online Mendelian Inheritance in Man, OMIM®. Johns Hopkins University, Baltimore, MD. MIM Number: {605713}: {2019 Nov 4}: World Wide Web URL: https://omim.org/
5 Online Mendelian Inheritance in Man, OMIM®. Johns Hopkins University, Baltimore, MD. MIM Number: {611120}: {2010 Sept 23}: World Wide Web URL: https://omim.org/
6 Penno A, Reilly MM, Houlden H, Laurá M, Rentsch K, Niederkofler V, Stoeckli ET, Nicholson G, Eichler F, Brown RH Jr, von Eckardstein A, Hornemann T. Hereditary sensory neuropathy type 1 is caused by the accumulation of two neurotoxic sphingolipids. J Biol Chem. 2010 Apr 9;285(15):11178-87. doi: 10.1074/jbc.M109.092973. Epub 2010 Jan 22. PMID: 20097765; PMCID: PMC2856995.
7 Rotthier A, Penno A, Rautenstrauss B, Auer-Grumbach M, Stettner GM, Asselbergh B, Van Hoof K, Sticht H, Lévy N, Timmerman V, Hornemann T, Janssens K. Characterization of two mutations in the SPTLC1 subunit of serine palmitoyltransferase associated with hereditary sensory and autonomic neuropathy type I. Hum Mutat. 2011 Jun;32(6):E2211-25. doi: 10.1002/humu.21481. Epub 2011 Feb 24. PMID: 21618344.
8 Auer-Grumbach M, Bode H, Pieber TR, Schabhüttl M, Fischer D, Seidl R, Graf E, Wieland T, Schuh R, Vacariu G, Grill F, Timmerman V, Strom TM, Hornemann T. Mutations at Ser331 in the HSN type I gene SPTLC1 are associated with a distinct syndromic phenotype. Eur J Med Genet. 2013 May;56(5):266-9. doi: 10.1016/j.ejmg.2013.02.002. Epub 2013 Feb 27. PMID: 23454272; PMCID: PMC3682180.
9Alecu I, Tedeschi A, Behler N, Wunderling K, Lamberz C, Lauterbach MA, Gaebler A, Ernst D, Van Veldhoven PP, Al-Amoudi A, Latz E, Othman A, Kuerschner L, Hornemann T, Bradke F, Thiele C, Penno A. Localization of 1-deoxysphingolipids to mitochondria induces mitochondrial dysfunction. J Lipid Res. 2017 Jan;58(1):42-59. doi: 10.1194/jlr.M068676. Epub 2016 Nov 23. PMID: 27881717; PMCID: PMC5234710.
10 Houlden H, King R, Blake J, Groves M, Love S, Woodward C, Hammans S, Nicoll J, Lennox G, O’Donovan DG, Gabriel C, Thomas PK, Reilly MM. Clinical, pathological and genetic characterization of hereditary sensory and autonomic neuropathy type 1 (HSAN I). Brain. 2006 Feb;129(Pt 2):411-25. doi: 10.1093/brain/awh712. Epub 2005 Dec 19. PMID: 16364956.
11 Reilly MM. Hereditary sensory neuropathy type 1 [Internet]. Danbury (CT): National Organization for Rare Disorders; 1991 [updated 2017; cited 2020 Nov 1]. Available from: https://rarediseases.org/rare-diseases/hereditary-sensory-neuropathy-type-i/
12 Nicholson GA. Hereditary Sensory Neuropathy Type IA. 2002 Sep 23 [Updated 2015 Sep 10]. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2018. https://www.ncbi.nlm.nih.gov/medgen/C0020071
13 Garofalo K, Penno A, Schmidt BP, Lee HJ, Frosch MP, von Eckardstein A, Brown RH, Hornemann T, Eichler FS. Oral L-serine supplementation reduces production of neurotoxic deoxysphingolipids in mice and humans with hereditary sensory autonomic neuropathy type 1. J Clin Invest. 2011 Dec;121(12):4735-45. doi: 10.1172/JCI57549. PMID: 22045570; PMCID: PMC3225995.
14 Gantner ML, Eade K, Wallace M, Handzlik MK, Fallon R, Trombley J, Bonelli R, Giles S, Harkins-Perry S, Heeren TFC, Sauer L, Ideguchi Y, Baldini M, Scheppke L, Dorrell MI, Kitano M, Hart BJ, Cai C, Nagasaki T, Badur MG, Okada M, Woods SM, Egan C, Gillies M, Guymer R, Eichler F, Bahlo M, Fruttiger M, Allikmets R, Bernstein PS, Metallo CM, Friedlander M. Serine and Lipid Metabolism in Macular Disease and Peripheral Neuropathy. N Engl J Med. 2019 Oct 10;381(15):1422-1433. doi: 10.1056/NEJMoa1815111. Epub 2019 Sep 11. PMID: 31509666.
15 Suh BC, Hong YB, Nakhro K, Nam SH, Chung KW, Choi BO. Early-onset severe hereditary sensory and autonomic neuropathy type 1 with S331F SPTLC1 mutation. Mol Med Rep. 2014 Feb;9(2):481-6. doi: 10.3892/mmr.2013.1808. Epub 2013 Nov 18. PMID: 24247255.
16 Fridman V, Suriyanarayanan S, Novak P, David W, Macklin EA, McKenna-Yasek D, Walsh K, Aziz-Bose R, Oaklander AL, Brown R, Hornemann T, Eichler F. Randomized trial of l-serine in patients with hereditary sensory and autonomic neuropathy type 1. Neurology. 2019 Jan 22;92(4):e359-e370. doi: 10.1212/WNL.0000000000006811. Epub 2019 Jan 9. PMID: 30626650; PMCID: PMC6345118.
17 Scherer SS. The debut of a rational treatment for an inherited neuropathy? J Clin Invest. 2011 Dec;121(12):4624-7. doi: 10.1172/JCI60511. PMID: 22045569; PMCID: PMC3226011.
18 https://neuromuscular.wustl.edu/time/hmsn.html
19 C.T. Hsiao, H. C. Chao, Y.C. Liao, K.P. Lin, B.W. Soong, Y.C. Lee, Investigation for SPTLC1 mutations in a Taiwanese cohort with hereditary neuropathies. Journal of the Neurological Science. 2017. https://doi.org/10.1016/j.jns.2017.08.3516
20 Thomas, P. K., and Ochoa, J. (1993) Clinical features and differential diagnosis. in Peripheral Neuropathy, 3rd Editio (Dyck, P. J., Thomas, K. P., Griffin, J. W., Low, P. A., and Poduslo, J. F. eds), pp. 749–774, Saunders Philladelphia